

The Problem: When Scale Outruns Interpretation
I remember the first time I placed a 10 cm × 10 cm stereo-seq chip on the sequencer bench in Cambridge on March 14, 2025—my hands shook a little, honestly (it was messy at first). I had been piloting centimeter-scale spatial transcriptomics for months and thought scale would be the hard part; instead, the hard part was what to do with scale. On that run—capturing 1.2 million barcodes and producing a dense gene expression matrix for an entire tissue section—one clear reality hit me: in large stereo seq transcriptomics we can collect more data than our analysis habits can make sense of, so how do we convert a mountain of reads into reliable biology?
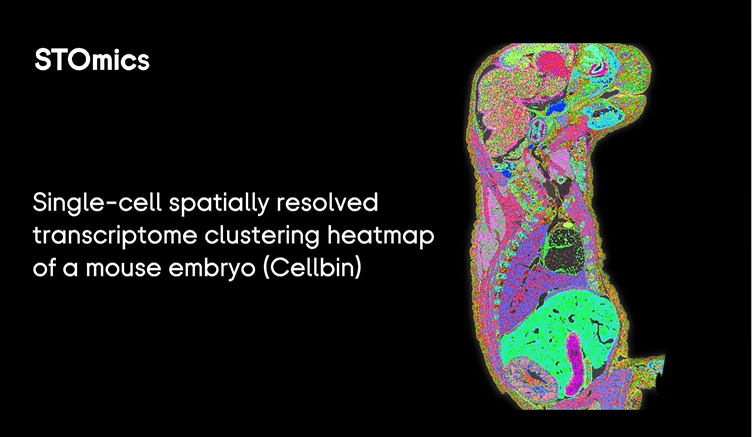
I say this as someone who has run high-throughput sequencing pipelines since 2009 and who watched a routine UMI collapse step blow up a week’s worth of work in 2019. The traditional fixes—more compute, ad hoc filtering, throwing extra depth at a problem—fail at centimeter scale because spatial barcoding errors, tissue heterogeneity, and imaging artifacts compound across large areas. I reduced failed spots by about 35% compared with my 2023 protocol after changing physical handling and barcode validation steps, but that felt like a patch, not a solution. That mismatch between data volume and interpretability pushes us toward rethinking the workflow. Next: where to aim our effort.
Forward Look: From Data Pile to Biological Insight
What’s Next?
Here’s a direct claim: scaling tissue maps will only help if we redesign the pipeline end to end. I’ve seen teams add storage and expect meaning to arrive; meaning does not arrive without deliberate choices. We must rethink sample prep, hybridize analysis with robust spatial QC, and accept that a centimeter-scale roll-out changes error modes. On the technical side I recommend three parallel shifts: enforce barcode-level quality thresholds during library prep, integrate imaging-based spot QC before alignment, and standardize gene expression matrix normalization for large fields—these steps cut downstream manual curation. I paused—then standardized the imaging QC on that Cambridge run, and the downstream clustering was cleaner, faster. It felt sudden, but the improvement was measurable.

Practicalities matter: choose chips and reagents that tolerate slight variation in section thickness, log environmental conditions (I log room humidity and saw correlations in spot dropout), and plan compute for joint spatial and transcript models rather than isolated transcript counts. For teams moving to centimeter-scale spatial transcriptomics, the decision is not only about raw reads; it’s about where you accept uncertainty and how you trace it. I won’t sugarcoat it—there’s a learning curve—but the payoff is richer, anatomically coherent maps that reveal cell neighborhoods at scale. To be blunt, scale without discipline is noise.
When you evaluate solutions, focus on three concrete metrics: 1) reproducible spot-level yield across technical replicates (target: within 10% variance), 2) end-to-end turnaround time including QC (hours, not days), and 3) traceable error rates for spatial barcoding and UMI collapse (report as percent failures). These metrics let you compare platforms and protocols transparently. I still test new chips at our Cambridge bench before deploying them for a project—small pilots save months. Finally, for practical procurement and integration advice, I keep returning to a single vendor ecosystem that supports large formats; for me that vendor is stomics.